mitochondria as central hub of pulmonary inflammation in cystic fibrosis
The ideal drug or treatment for cystic fibrosis (CF) should be one that rescues and potentiates the residual functionality of defective CF Transmembrane conductance Regulator (CFTR) channel, but also generates favourable conditions to interrupt the vicious circle that sustains lung hyperinflammation generated by chronic bacterial infection and defective CFTR-dependent intracellular signaling.
In this regard, we have demonstrated that abnormal mitochondrial calcium (Ca2+) signaling exacerbates the airway inflammatory responses and affects the autophagy in CF [1,2]. Among the defects associated to CFTR channel, the perturbed Ca2+ signaling has a relevant role in the pathophysiology of CF. The Ca2+ signaling dysregulation has been observed in several human CF patient-derived primary cells, including airway epithelial and immune cells [1,3-5] (Figure 1).
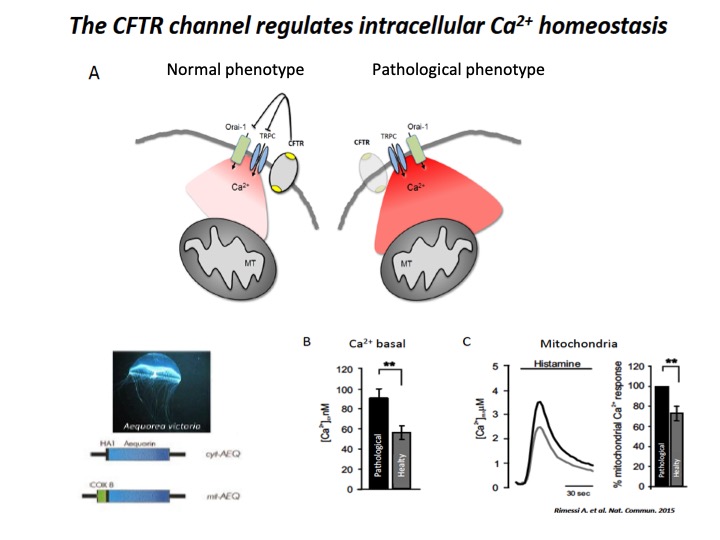
Fig. 1: Implication of CFTR in Ca2+ signaling
A) Schematic representation of intracellular Ca2+ overload in Cystic fibrosis airway cells (pathological phenotype). The cellular Ca2+ homeostasis is regulated by systems of Ca2+-entry and Ca2+-efflux located in plasma membrane and organelles, which are the principal molecular systems involved in the abnormal intracellular Ca2+ signaling associated with defective CFTR channel, where their dysfunction contribute to physiopathology of CF lung disease. B) Basal [Ca2+] ± ES in CF (pathological) vs non-CF (healthy) cells under resting conditions. C) Representative histamine-dependent mitochondrial Ca2+ response in pathological and healthy cells. The histograms show the % change of the Ca2+ responses in control compared with CF cells.
In all of them, the mitochondrial Ca2+ concentration was increased compared to non-CF cells, with heavy repercussions about the susceptibility to pathogens, which led to mitochondrial dysfunction and autophagy impairment, resulting in persistent exacerbations, defective bacterial uptake, killing and clearance and excessive cytokines release in CF lung [6-10] (Figure 2).
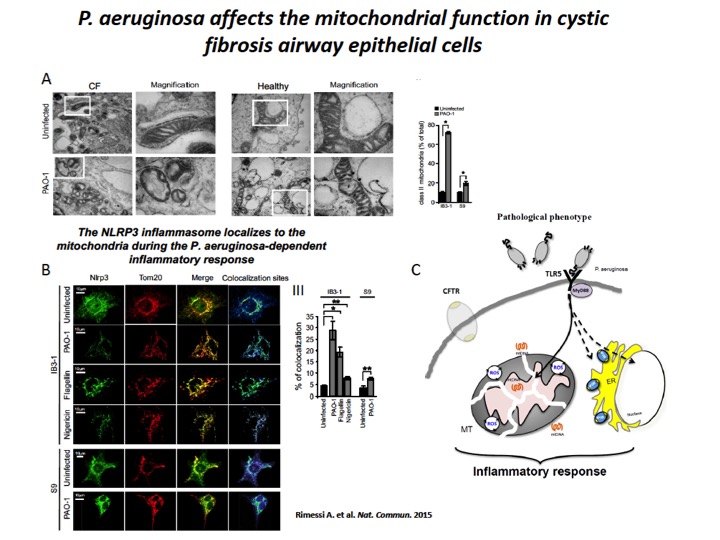
Fig. 2: P. aeruginosa induces mitochondrial dysfunction and NLRP3 activation in CF airway cells
A) The mitochondria in CF airway cells show higher susceptibility to P. aeruginosa-dependent structural perturbations than non-CF cells. Representative transmission EM fields of mitochondria of IB3-1 and S9 cells treated with the PAO-1 strain are shown (× 10,000). The white box represents × 80,000 magnification of a single mitochondrion. Morphometric analysis of randomly selected EM fields of mitochondria class II in IB3-1 and S9 cells. The data represent the % of mitochondrial class II±s.e. in reference to the total mitochondria of three independent experiments. B) P. aeruginosa induces mitochondrial NLRP3 localization and activation in CF airway cells. Confocal immunofluorescence images of IB3-1 and S9 cells endogenously expressing NLRP3. Co-localization of NLRP3 with the mitochondrial protein TOM20 during the PAO-1-, flagellin- and nigericin-dependent responses. IB3-1 cells were treated with PAO-1 (100 MOI) or flagellin (10 μg ml−1) for 2 h and with nigericin (10 μM) for 30 min. Quantification of NLRP3 co-localization with mitochondria in IB3-1 cells, expressed as the % of co-localization, which was calculated as average volume of the overlapping areas. C) Schematic representation of P. aeruginosa-triggered mitochondrial damage-associated molecular patterns (mtDAMPs) releases and consequent inflammatory activation in CF airway cells, due to mitochondrial dysfunction.
Typically, the mitochondria were considered as a central signaling hub to regulate specific functions, such as metabolism and/or cell death, through the regulation of Ca2+ homeostasis mediating interorganelle communication, in particular with ER (Figure 3). Now, continue evidence indicate that the mitochondria act as central regulator, checkpoint and arbitrator also for the inflammatory process. Mitochondrial dysfunction and altered Ca2+ signaling sustain inflammation in different inflammatory-related pathological conditions, including CF.
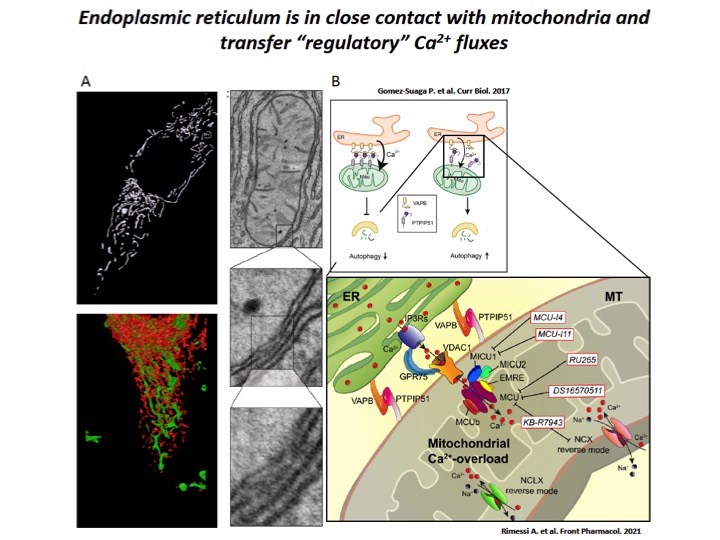
Fig. 3: Dampening the mitochondrial Ca2+-overload in cystic fibrosis
A) ER-mitochondria interfaces are intimate and dynamic regions between ER and mitochondrial outer membranes, where a series of specialized molecular bridges control the frequency of interactions, the size and the spacing between the organelles, changing at front of cellular and functional requests. High-resolution 3D imaging of mitochondria (green) and ER (red) in a HeLa cell and representative transmission EM fields of ER-mitochondria juxtaposition. B) During the recurrent pathogen infections, the increased of ER-mitochondria juxtaposition induces the dysregulation of Ca2+ signaling in CF, which causes mitochondrial Ca2+-overload in airway cells with consequent autophagic impairment and inflammation. The mitochondrial Ca2+-overload is mediated by an increased ER-mitochondria Ca2+ transfer through the IP3Rs-VDAC-MCU axis due to the stabilization of VAPB-PTPIP51 tethers. Indeed, the increased ENaC-dependent Na+ absorption due to defective CFTR in CF could stimulate NCX and NCLX exchangers to work in reverse mode triggering intracellular and mitochondrial Ca2+-influx, which may worsen the excessive mitochondrial Ca2+-uptake. To dampen the detrimental Ca2+ accumulation in matrix, a new class of mitochondrial Ca2+ modulator drugs are under investigation.
In these years, we have demonstrated that in CF airway epithelial cells, during P. aeruginosa infection, increased the ER-mitochondria juxtapositions due by stabilization of ER protein vesicle-associated membrane protein-associated protein B and the outer mitochondrial membrane protein tyrosine phosphatase interacting protein 51 tethers, favoring the ER-mitochondria Ca2+ transfer, via Mitochondrial Calcium Uniporter (MCU) [2] (Figure 4). This led to organelle dysfunction in CF airways cells, with consequent mitochondrial membrane potential alteration and radical oxygen species (ROS) production, inducing persistent mitochondrial Unfolding Protein Response (UPRmt) and NLRP3 inflammasome activation. In turn, these processes downregulated the selective autophagic responses, mitophagy (selective degradation of altered mitochondria by autophagy) and xenophagy (selective removal of intracellular pathogens by autophagy), resulting in augmented pathogen survival and worsening of inflammatory response in CF lung [1,2] (Figure 4).
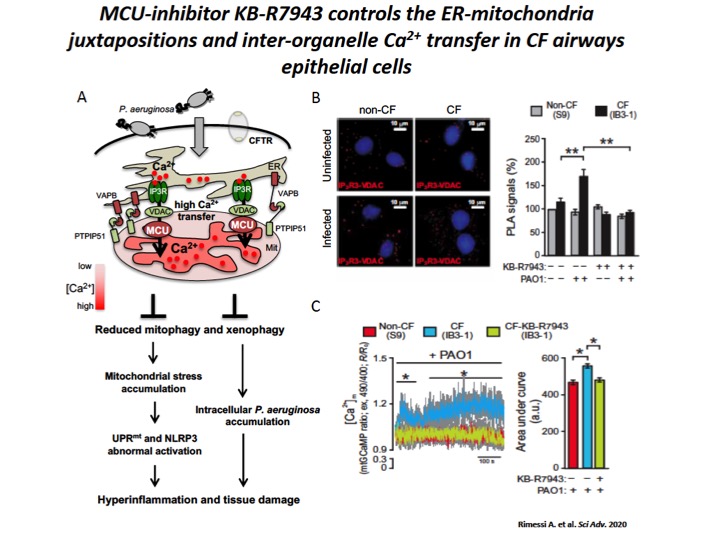
Fig. 4: The increase of ER-mitochondria tethering facilitates the inter-organelle Ca2+ transfer, via MCU, in CF airway cells during P. aeruginosa infection.
A) Schematic representation of increased inter-organelle Ca2+ transfer due by VAPB and PTPIP51 tethering stabilization in CF airway cells during P. aeruginosa infection, with relative consequences associated to selective autophagy impairments. Reduced selective autophagic responses (mitophagy and xenophagy) potentiate UPRmt and inflammasome activation, favoring a vicious cycle that sustain the hyperinflammation in CF. B) Representative images with Protein Ligation Assay (PLA) signals (red) in the different cells are shown. The cell nuclei were stained with 4′,6-diamidino-2-phenylindole (blue). The bar chart shows quantification of IP3-R3-VDAC PLA signals (%), respect to uninfected non-CF cells. C) Mitochondrial Ca2+ dynamics in S9- (non-CF), IB3-1– (CF), and KB-R7943–treated IB3-1 (CF-KB-R7943) cells exposed to PAO1 at an MOI of 100 were evaluated through ratiometric imaging of mitochondrial-targeted GCaMP6.
Controlling the mitochondrial Ca2+-overload, via KB-R7943-dependent MCU inhibition, we attenuated in vitro and in vivo the P. aeruginosa-dependent mitochondrial dysfunction and hyperinflammation in CF lung, controlling UPRmt and NLRP3 inflammasome activation, while the selective autophagic responses were rectified (Figure 5) [2]. A series of results that have provided the proof-of-concept that the mitochondrial Ca2+-overload in CF may be modulated to delay the onset of CF lung disease and to influence the inflammation and autophagy.
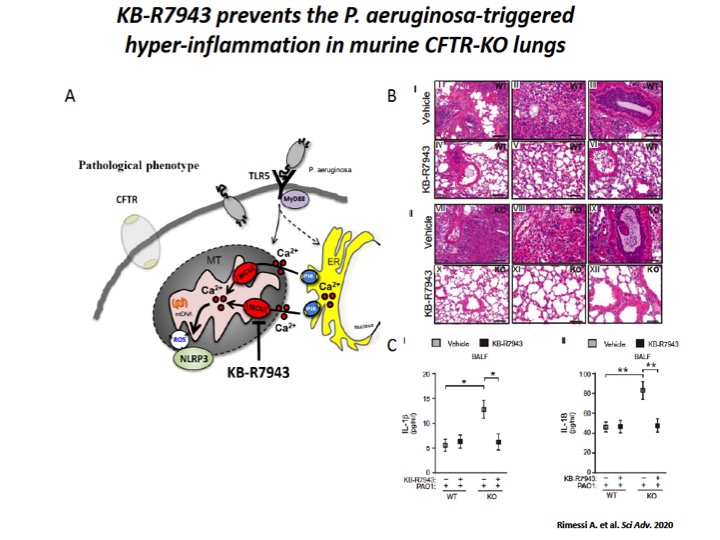
Fig. 5: The mitochondrial Ca2+ modulator, KB-R7943, limits the P. aeruginosa–triggered inflammatory response in vivo.
A) Schematic representation in which KB-R7943, inhibiting MCU, limits the activation of NLRP3 inflammasome in CF airways through the regulation of inter-organelle Ca2+ transfer via MCU. B) Reduced degree and extension of inflammation in CF lungs when pretreated with mitochondrial Ca2+ modulator, KB-R7943. The images exemplify the lungs of WT and CFTR-KO mice stained with hematoxylin and eosin (H&E), pretreated 1 hour before infection with KB-R7943 and vehicle. Scale bars, 250 μm (I, IV, VII, and X) and 500 μm (II, III, V, VI, VIII, IX, XI, and XII). C) The levels of released IL-1β (I) and IL-18 (II) measured in BALF of WT and CFTR-KO mice exposed to P. aeruginosa infection, treated with KB-R7943 and vehicle.
The main goal of our project is to gain pre-clinical insights in vitro and in vivo about a new class of safety and selective mitochondrial Ca2+-targeting agents that aim to control the mitochondrial Ca2+-overload and the autophagic impairment in CF airway epithelial and immune cells to prevent the exacerbation of inflammation and to cooperate with CFTR-modulators to ameliorate the restore of CFTR functioning in CF lung. We aim to determinate the anti-inflammatory effects of mitochondrial Ca2+-modulators in vitro in CF patient-derived airway epithelial cells and in vivo in a murine CFTR-null and DF508-CFTR models. We will determinate whether mitochondrial Ca2+-modulators, acting also as autophagy modulating compounds, cooperate with CFTR-modulators (CFTR correctors and potentiators) to improve the residue functionality of defective CFTR. Finally, we will determinate whether mitochondrial Ca2+-modulators, safeguarding the mitochondrial physiology and controlling the autophagy, influence the perturbed polarization reprogramming and antimicrobial response of CF immune cells, considering that the hyperinflammation observed in CF lung is in part conditioned by altered phenotype of airway immune cells associated to defective CFTR.
We expect that controlling mitochondrial Ca2+ signaling will counteract the amplification of inflammatory signals in CF airway and immune cells during pathogen infection, controlling the mitochondrial function.
References
1. Rimessi A, Bezzerri V, Patergnani S, Marchi S, Cabrini G, Pinton P. Mitochondrial Ca2+ -dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat Commun. 2015;6:6201.
2. Rimessi A, Pozzato C, Carparelli L, Rossi A, Ranucci S, De Fino I, Cigana C, Talarico A, Wieckowski MR, Ribeiro CMP, Trapella C, Rossi G, Cabrini G, Bragonzi A, Pinton P. Pharmacological modulation of mitochondrial calcium uniporter controls lung inflammation in cystic fibrosis. Sci Adv. 2020;6:eaax9093.
3. Robledo-Avila FH, Ruiz-Rosado JD, Brockman KL, Kopp BT, Amer AO, McCoy K, Bakaletz LO, Partida-Sanchez S. Dysregulated Calcium Homeostasis in Cystic Fibrosis Neutrophils Leads to Deficient Antimicrobial Responses. J Immunol. 2018;201:2016-2027.
4. Waller RL, Brattin WJ, Dearborn DG. Cytosolic free calcium concentration and intracellular calcium distribution in lymphocytes from cystic fibrosis patients. Life Sci. 1984;35:775-81.
5. Rimessi, A., Vitto, V. A. M., Patergnani, S. and Pinton, P. Update on calcium signaling in cystic fibrosis lung disease. Front in Pharmacol 2021
6. Brockman SM, Bodas M, Silverberg D, Sharma A, Vij N. Dendrimer-based selective autophagy-induction rescues ΔF508-CFTR and inhibits Pseudomonas aeruginosa infection in cystic fibrosis. PLoS One. 2017;12:e0184793.
7. Ferrari E, Monzani R, Villella VR, Esposito S, Saluzzo F, Rossin F, D’Eletto M, Tosco A, De Gregorio F, Izzo V, Maiuri MC, Kroemer G, Raia V, Maiuri L. Cysteamine re-establishes the clearance of Pseudomonas aeruginosa by macrophages bearing the cystic fibrosis-relevant F508del-CFTR mutation. Cell Death Dis. 2017;8:e2544.
8. Pehote G, Bodas M, Brucia K, Vij N. Cigarette Smoke Exposure Inhibits Bacterial Killing via TFEB-Mediated Autophagy Impairment and Resulting Phagocytosis Defect. Mediators Inflamm. 2017;2017:3028082.
9. Shrestha CL, Assani KD, Rinehardt H, Albastroiu F, Zhang S, Shell R, Amer AO, Schlesinger LS, Kopp BT. Cysteamine-mediated clearance of antibiotic-resistant pathogens in human cystic fibrosis macrophages. PLoS One. 2017;12:e0186169.
10. Patergnani S, Vitto VAM, Pinton P, Rimessi A. Mitochondrial Stress Responses and “Mito-Inflammation” in Cystic Fibrosis. Front Pharmacol. 2020;11:581114.